Function for rendering sequence index plots with ggplot2
instead of base R's plot function that is used by
TraMineR::seqrfplot. Note that ggseqrfplot
uses patchwork to combine the different components of
the plot. The function and the documentation draw heavily from
TraMineR::seqrf.
Usage
ggseqrfplot(
seqdata = NULL,
diss = NULL,
k = NULL,
sortv = "mds",
weighted = TRUE,
grp.meth = "prop",
squared = FALSE,
pow = NULL,
seqrfobject = NULL,
border = FALSE,
ylab = NULL,
yaxis = TRUE,
which.plot = "both",
quality = TRUE,
box.color = NULL,
box.fill = NULL,
box.alpha = NULL,
outlier.jitter.height = 0,
outlier.color = NULL,
outlier.fill = NULL,
outlier.shape = 19,
outlier.size = 1.5,
outlier.stroke = 0.5,
outlier.alpha = NULL
)Arguments
- seqdata
State sequence object (class
stslist) created with theTraMineR::seqdeffunction.seqdatais ignored ifseqrfobjectis specified.- diss
pairwise dissimilarities between sequences in
seqdata(seeTraMineR::seqdist).dissis ignored ifseqrfobjectis specified.- k
integer specifying the number of frequency groups. When
NULL,kis set as the minimum between 100 and the sum of weights over 10.kis ignored ifseqrfobjectis specified.- sortv
optional sorting vector of length
nrow(diss)that may be used to compute the frequency groups. IfNULL, the original data order is used. Ifmds(default), the first MDS factor ofdiss(diss^2whensquared=TRUE) is used. Ties are randomly ordered. Also allows for the usage of the string inputs:"from.start"or"from.end"(seeggseqiplot).sortvis ignored ifseqrfobjectis specified.- weighted
Controls if weights (specified in
TraMineR::seqdef) should be used. Default isTRUE, i.e. if available weights are used.- grp.meth
Character string. One of
"prop","first", and"random". Grouping method. See details.grp.methis ignored ifseqrfobjectis specified.- squared
Logical. Should medoids (and computation of
sortvwhen applicable) be based on squared dissimilarities? (default isFALSE).squaredis ignored ifseqrfobjectis specified.- pow
Dissimilarity power exponent (typically 1 or 2) for computation of pseudo R2 and F. When
NULL, pow is set as 1 whensquared = FALSE, and as 2 otherwise.powis ignored ifseqrfobjectis specified.- seqrfobject
object of class
seqrfgenerated withTraMineR::seqrf. Default isNULL; eitherseqrfobjectorseqdataanddisshave to specified- border
if
TRUEbars of index plot are plotted with black outline; default isFALSE(also acceptsNULL)- ylab
character string specifying title of y-axis. If
NULLaxis title is "Frequency group"- yaxis
Controls if a y-axis is plotted. When set as
TRUE, index of frequency groups is displayed.- which.plot
character string specifying which components of relative frequency sequence plot should be displayed. Default is
"both". If set to"medoids"only the index plot of medoids is shown. If"diss.to.med"only the box plots of the group-specific distances to the medoids are shown.- quality
specifies if representation quality is shown as figure caption; default is
TRUE- box.color
specifies color of boxplot borders; default is "black
- box.fill
specifies fill color of boxplots; default is "white"
- box.alpha
specifies alpha value of boxplot fill color; default is 1
- outlier.jitter.height
if greater than 0 outliers are jittered vertically. If greater than .375 height is automatically adjusted to be aligned with the box width.
- outlier.color, outlier.fill, outlier.shape, outlier.size, outlier.stroke, outlier.alpha
parameters to change the appearance of the outliers. Uses defaults of
ggplot2::geom_boxplot
Value
A relative frequency sequence plot using ggplot.
Details
This function renders relative frequency sequence plots using either an internal
call of TraMineR::seqrf or by using an object of
class "seqrf" generated with TraMineR::seqrf.
For further details on the technicalities we refer to the excellent documentation
of TraMineR::seqrf. A detailed account of
relative frequency index plot can be found in the original contribution by
Fasang and Liao (2014)
.
ggseqrfplot renders the medoid sequences extracted by
TraMineR::seqrf with an internal call of
ggseqiplot. For the box plot depicting the distances to the medoids
ggseqrfplot uses geom_boxplot and
geom_jitter. The latter is used for plotting the outliers.
Note that ggseqrfplot renders in the box plots analogous to the those
produced by TraMineR::seqrfplot. Actually,
the box plots produced with TraMineR::seqrfplot
and ggplot2::geom_boxplot
might slightly differ due to differences in the underlying computations of
grDevices::boxplot.stats and
ggplot2::stat_boxplot.
Note that ggseqrfplot uses patchwork to combine
the different components of the plot. If you want to adjust the appearance of
the composed plot, for instance by changing the plot theme, you should consult
the documentation material of patchwork.
At this point ggseqrfplot does not support a grouping option. For
plotting multiple groups, I recommend to produce group specific seqrfobjects or
plots and to arrange them in a common plot using patchwork.
See Example 6 in the vignette for further details:
vignette("ggseqplot", package = "ggseqplot")
References
Fasang AE, Liao TF (2014). “Visualizing Sequences in the Social Sciences: Relative Frequency Sequence Plots.” Sociological Methods & Research, 43(4), 643–676. doi:10.1177/0049124113506563 .
Examples
library(TraMineR)
library(ggplot2)
library(patchwork)
# From TraMineR::seqprf
# Defining a sequence object with the data in columns 10 to 25
# (family status from age 15 to 30) in the biofam data set
data(biofam)
biofam.lab <- c("Parent", "Left", "Married", "Left+Marr",
"Child", "Left+Child", "Left+Marr+Child", "Divorced")
# Here, we use only 100 cases selected such that all elements
# of the alphabet be present.
# (More cases and a larger k would be necessary to get a meaningful example.)
biofam.seq <- seqdef(biofam[501:600, 10:25], labels=biofam.lab,
weights=biofam[501:600,"wp00tbgs"])
#> [>] 8 distinct states appear in the data:
#> 1 = 0
#> 2 = 1
#> 3 = 2
#> 4 = 3
#> 5 = 4
#> 6 = 5
#> 7 = 6
#> 8 = 7
#> [>] state coding:
#> [alphabet] [label] [long label]
#> 1 0 0 Parent
#> 2 1 1 Left
#> 3 2 2 Married
#> 4 3 3 Left+Marr
#> 5 4 4 Child
#> 6 5 5 Left+Child
#> 7 6 6 Left+Marr+Child
#> 8 7 7 Divorced
#> [>] sum of weights: 111.62 - min/max: 0/4.17260217666626
#> [>] 100 sequences in the data set
#> [>] min/max sequence length: 16/16
diss <- seqdist(biofam.seq, method = "LCS")
#> [>] 100 sequences with 8 distinct states
#> [>] creating a 'sm' with a substitution cost of 2
#> [>] creating 8x8 substitution-cost matrix using 2 as constant value
#> [>] 76 distinct sequences
#> [>] min/max sequence lengths: 16/16
#> [>] computing distances using the LCS metric
#> [>] elapsed time: 0.017 secs
# Using 12 groups and default MDS sorting
# and original method by Fasang and Liao (2014)
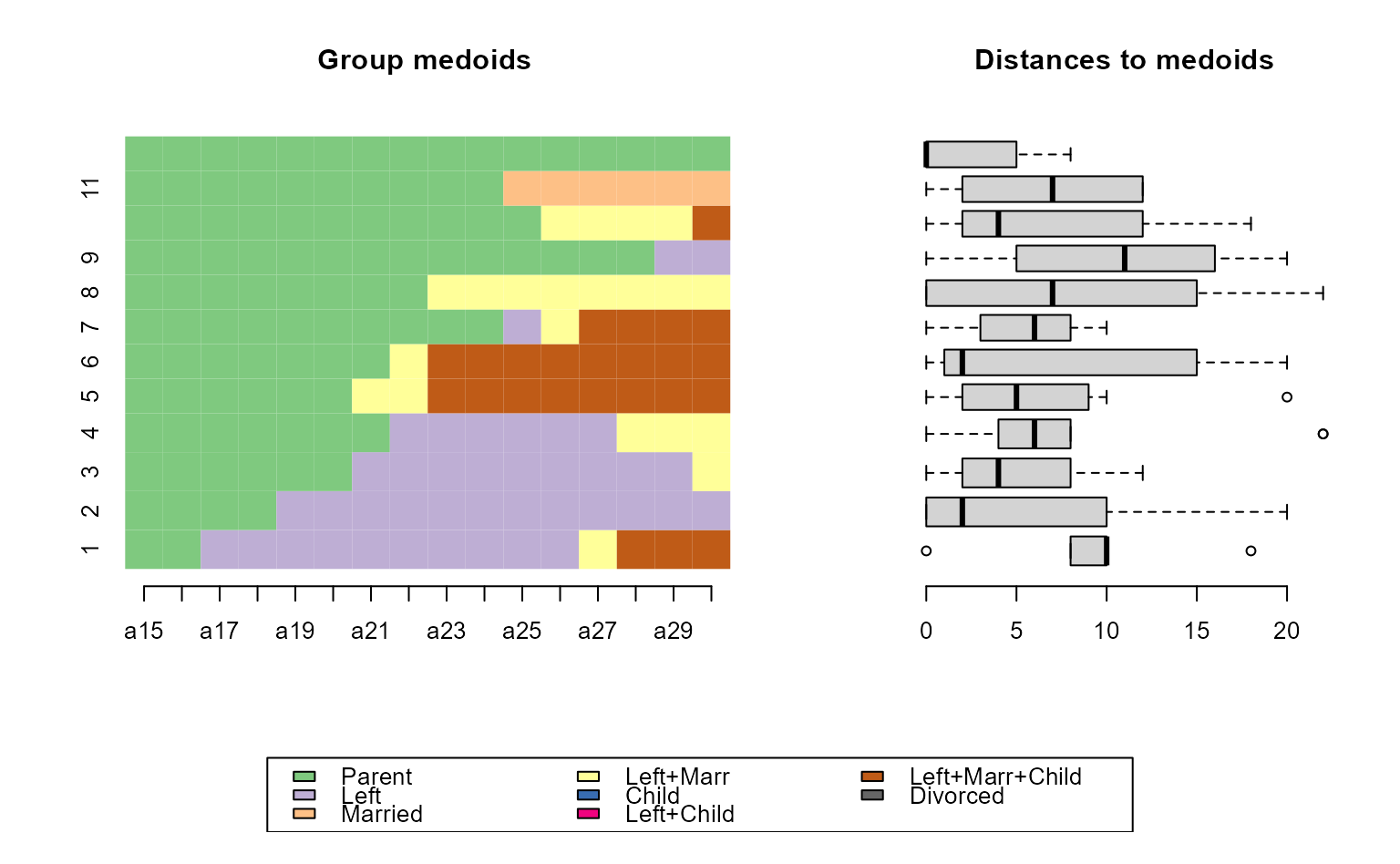
# ... with TraMineR::seqrfplot (weights have to be turned off)
seqrfplot(biofam.seq, weighted = FALSE, diss = diss, k = 12,
grp.meth="first", which.plot = "both")
#> [>] Using k=12 frequency groups with grp.meth='first'
#> [>] Pseudo/medoid-based-R2: 0.4620155
#> [>] Pseudo/medoid-based-F statistic: 6.870317, p-value: 3.09994e-08
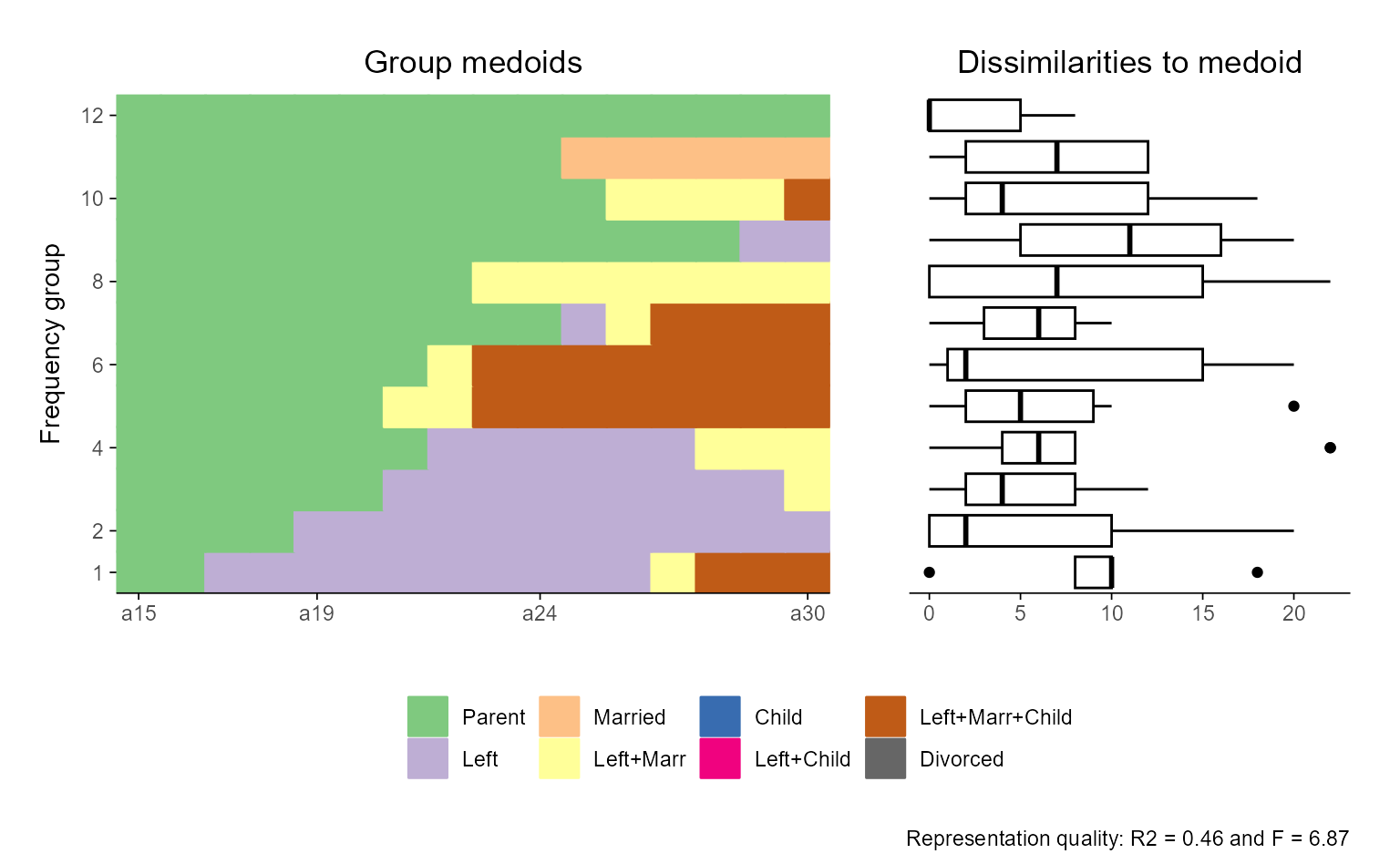
 # ... with ggseqrfplot
ggseqrfplot(biofam.seq, weighted = FALSE, diss = diss, k = 12, grp.meth="first")
#> [>] Using k=12 frequency groups with grp.meth='first'
#> [>] Pseudo/medoid-based-R2: 0.4620155
#> [>] Pseudo/medoid-based-F statistic: 6.870317, p-value: 3.09994e-08
# ... with ggseqrfplot
ggseqrfplot(biofam.seq, weighted = FALSE, diss = diss, k = 12, grp.meth="first")
#> [>] Using k=12 frequency groups with grp.meth='first'
#> [>] Pseudo/medoid-based-R2: 0.4620155
#> [>] Pseudo/medoid-based-F statistic: 6.870317, p-value: 3.09994e-08
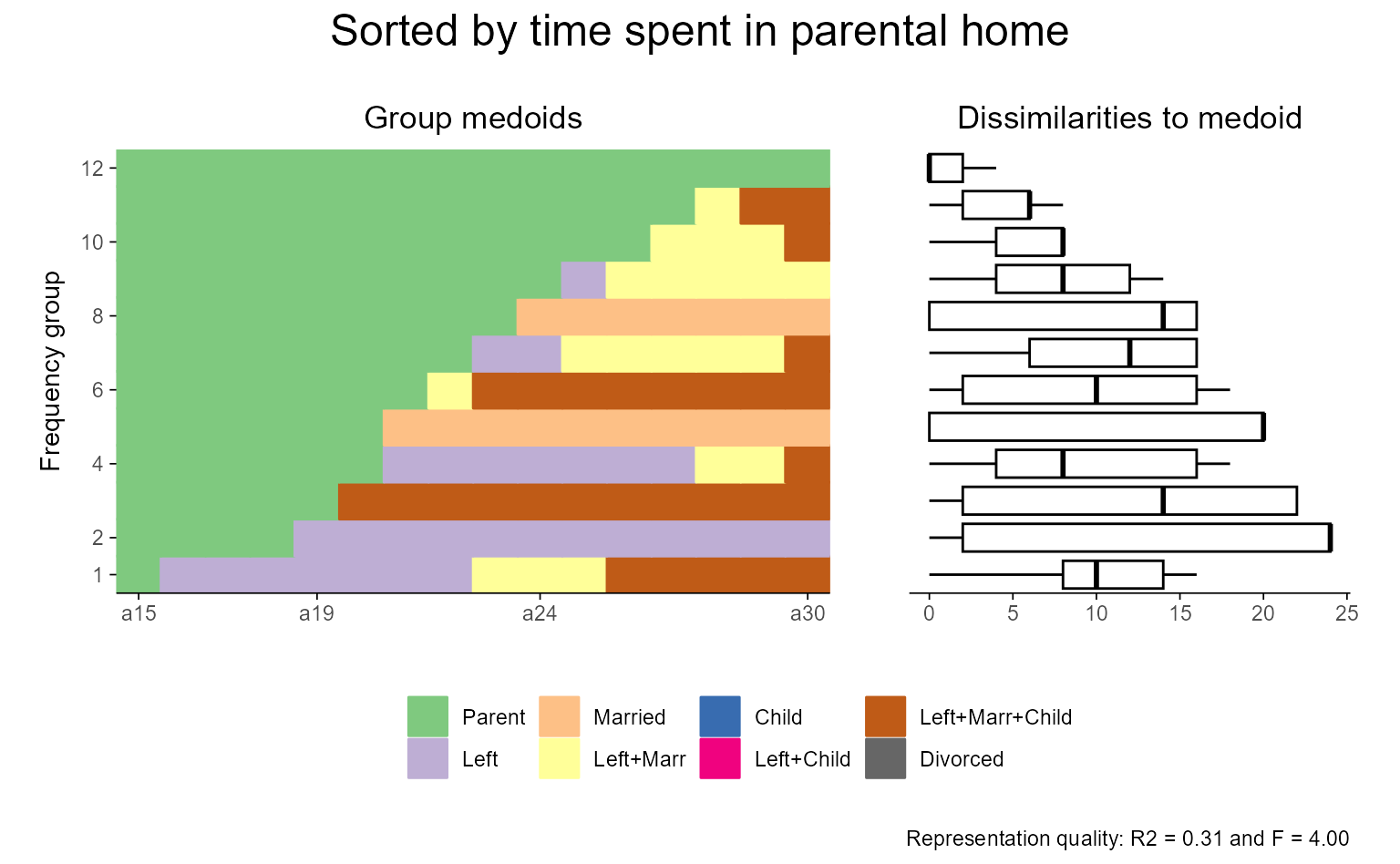
 # Arrange sequences by a user specified sorting variable:
# time spent in parental home; has ties
parentTime <- seqistatd(biofam.seq)[, 1]
#> [>] computing state distribution for 100 sequences ...
b.srf <- seqrf(biofam.seq, diss=diss, k=12, sortv=parentTime)
#> [>] Using k=12 frequency groups with grp.meth='prop'
#> [>] Pseudo/medoid-based-R2: 0.3064171
#> [>] Pseudo/medoid-based-F statistic: 4.001018, p-value: 7.736543e-05
# ... with ggseqrfplot (and some extra annotation using patchwork)
ggseqrfplot(seqrfobject = b.srf) +
plot_annotation(title = "Sorted by time spent in parental home",
theme = theme(plot.title = element_text(hjust = 0.5, size = 18)))
# Arrange sequences by a user specified sorting variable:
# time spent in parental home; has ties
parentTime <- seqistatd(biofam.seq)[, 1]
#> [>] computing state distribution for 100 sequences ...
b.srf <- seqrf(biofam.seq, diss=diss, k=12, sortv=parentTime)
#> [>] Using k=12 frequency groups with grp.meth='prop'
#> [>] Pseudo/medoid-based-R2: 0.3064171
#> [>] Pseudo/medoid-based-F statistic: 4.001018, p-value: 7.736543e-05
# ... with ggseqrfplot (and some extra annotation using patchwork)
ggseqrfplot(seqrfobject = b.srf) +
plot_annotation(title = "Sorted by time spent in parental home",
theme = theme(plot.title = element_text(hjust = 0.5, size = 18)))